My research broadly focuses on virus evolution, genome evolution, and phylogenetics; with special attention to virus-host co-evolution, the evolution and origin of specific genes, and phenotypic consequences of genotypic novelty. Viruses are obligate intracellular parasites infecting organisms across the three domains of life (Archaea, Bacteria and Eukarya). New viruses are continuously being discovered and it has become apparent that viruses are virtually everywhere. Viruses are important drivers of ecosystem functioning, as viral infection can influence the evolution of their hosts as well as that of other organisms (including other viruses) interacting with these hosts. Due to the different lifestyles, there is astounding variation in genome architecture (i.e. genome organization) evolution between viral families, but also between closely related viruses. To better understand how viruses co-evolve with their hosts, and how viruses evolve specific genome architectures, I combine both experimental and computational methods. I use computational molecular evolution in combination with comparative genomics to better understand the phylogenetic relationships among viruses, to reconstruct large insertion/deletion/recombination events, and to test the evolution and origin of specific genes and genomes. I use experimental evolution to follow fitness and genome adaptation of viruses over time under varying conditions. During these evolution experiments, I asses viral fitness by means of accumulation and within-host competitive fitness experiments. Changes in the viral genome (including signs of adaptation) are assessed by performing next-generation sequencing. I use other techniques such as RNA-sequencing and proteomics to asses more specific questions. For example, whether the same virus uses different sets of genes in different hosts, or whether the host response to infection is different under varying environmental conditions. In addition to experimental evolution, I use different sequencing methods for the discovery of novel viruses.
Host range and genome adaptation of giant viruses
For more than a century, viruses were considered tiny particles, fully dependent on their host cells to replicate. The recent discovery of giant viruses, containing unusually large genomes, challenged this assumption and blurred the sharp division between viruses and cellular life. It was also striking to learn that many of the giant virus genomes encode translation-related genes, indicating that they are presumably more independent -in terms of translation- as compared to other viruses and potentially infect and replicate in a broad range of hosts. Nonetheless, for most of the giant viruses, the precise host range and natural host species remain to be investigated. In the proposed project, I will investigate the host range and genome adaptation of giant viruses by combining in silico and wet-lab strategies. I postulate that, codon usage is an important factor in the adaptation of giant viruses to their hosts, where well-adapted codon usage provides for superior viral fitness. I will analyse the codon usage preferences among giant viruses and correlate these with the known and presumable hosts they infect. This will allow me to computationally predict the best-suited hosts, and subsequently, experimentally assess my predictions on different laboratory hosts. To further investigate whether codon usage defines the rate of genome adaptation of giant viruses, I will perform experimental evolution over a half year time period. The origin and evolutionary history of giant viruses is controversially discussed. By investigating the evolutionary relationships between giant viruses and their hosts in the context of their codon usage preferences, I will contribute to a better understanding of the factors determining host range and the evolutionary processes shaping giant virus genomes. Disentangling the connection between genomic content and host range will provide important knowledge in the fields of virology, evolutionary biology, genomics, and virus-host interactions.
This project is financed by a Marie Skłodowska-Curie Postdoctoral Fellowship (H2020-MSCA-IF-2019 GIVIREVOL, Grant agreement 891572) and the University of Vienna.
The evolutionary history of oncogenic and non-oncogenic papillomaviruses
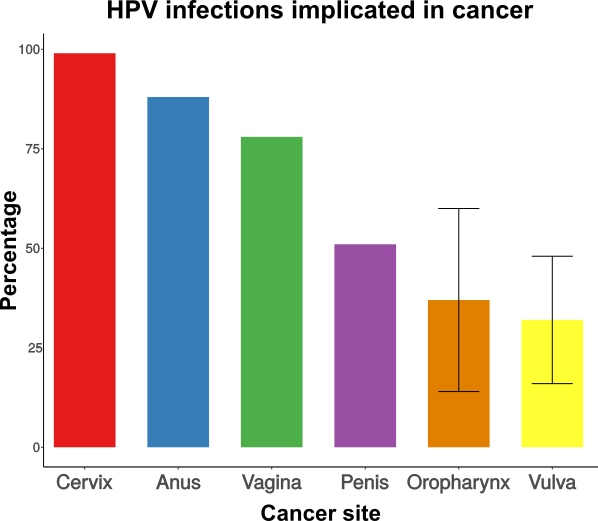
Papillomaviruses (PVs) have a wide host range, infecting mammals, birds, turtles, and snakes. Certain PVs are a major public health concern as in humans they are responsible for virtually all cases of cervical and anal cancer, and for a fraction of cancers on the penis, vagina, vulva and oropharynx (WHO Weekly Epidemiological Record 92:241-268 2017). But oncogenic PVs are actually an unfortunate exception, as most PVs cause asymptomatic infections, and a few cause benign, wart-like lesions. Despite the efforts directed towards the understanding of the different clinical manifestations of infection, our knowledge on PV evolution remains fragmentary. In this study I combine computational and experimental approaches to better understand the factors that differentiate between non-oncogenic and oncogenic PVs.
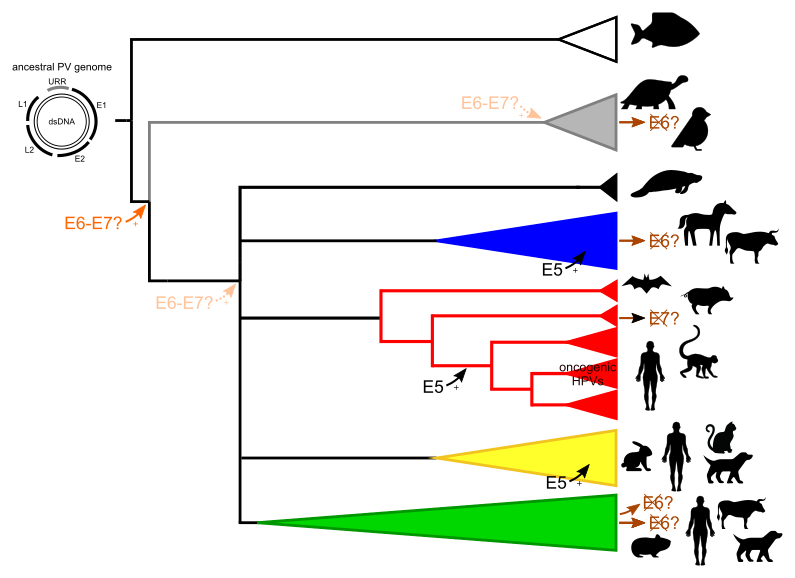
By using phylogenetic and comparative genomic methods, I started with a global analysis on the evolutionary history of the viral family. I have paid special attention to the PV oncogenes, which have followed different evolutionary histories than the PV backbone. The major oncoproteins, E6 and E7, are know to degrade tumor suppressor proteins which can eventually lead to the development of cancer. There exists also a minor oncoprotein, termed E5, of which the functions and origin remain to be fully elucidated. The recent discovery of PVs in fish suggests that ancestral PVs consisted of the minimal PV backbone (placing a new root on the phylogenetic tree) and that the E5, E6 and E7 oncogenes were acquired later during the evolution of PVs. These genes are directly involved in the onset of cancer, however, while almost all PV lineages contain at least E6 and/or E7, only a few lineages are responsible for cancer.
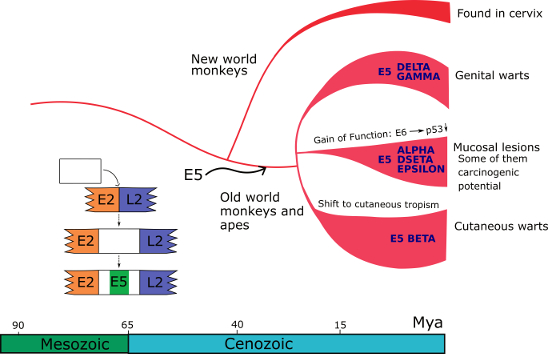
Bayesian phylogenetic analyses date the most recent common ancestor of the PV backbone to ~424 million years ago (Mya) (Willemsen and Bravo 2019). Common ancestry tests on extant E6 and E7 genes indicate that they share a common ancestor dating back to at least 184 Mya. The E5 genes, however, do not appear to share a common ancestor, but rather evolved de novo in a non-coding region in the genomes of a few polyphyletic PV lineages (Willemsen et. al. 2019). The entrance of E5 in the ancestral genome of PVs infecting primates concurred with an event that was instrumental for the differential oncogenic potential of present-day PVs infecting humans. During this adaptive radiation, certain E6 and E7 proteins acquired the ability to degrade tumor suppressor proteins (p53 and pRB) and facilitate the development of cancer in different tissues.
To further understand the connection between genotype and phenotype, we have resurrected the ancestral sequences of E6 and E7 by means of phylogenetic methods. Currently, we are experimentally testing the properties of both extant and ancestral genes in human cell lines. Preliminary results suggest that different alternative spliced mRNAs exist, which are thought to be related to the oncogenicity of certain PVs. We are currently performing total proteomics and transcriptomics to assess the impact of viral protein expression on cellular function. In addition, in collaboration with the Gilles Trave lab, we are testing the binding affinity of the ancestral E6 proteins to specific motifs that mediate or are related to the degradation of tumor suppressor proteins, and thus the onset of cancer.
This project was financed by a Marie Skłodowska-Curie Postdoctoral Fellowship (H2020-MSCA-IF-2016 ONCOGENEVOL, Grant agreement 750180) and developed during my first (and a half) postdoctoral stay working for the CNRS at the "Infectious Diseases and Vectors: Ecology, Genetics, Evolution and Control" (MIVEGEC) institute.
Evolution of viral codon usage preferences: manipulation of translation accuracy and evasion of immune response
In my previous lab, I also contributed to an ERC project that studies the evolution of viral codon usage preferences. In this project I contributed to two specific aims. The first aim was to analyze the impact of codon usage preferences in synonymous genes using a cellular model. To do this we monitored the differential evolution of a set of synonymous genes encoding for antibiotic resistance by means of experimental evolution of transformed cells in culture. The second aim was to analyze the impact of codon usage on the virus-cell interplay. To do this we quantified (and are still quantifying) the differential impact on protein fitness of a series of synonymous versions of the extant E6 and E7 viral oncogenes with transforming activity.
This project was financed by an ERC Consolidator Grant to Ignacio G Bravo (H2020-ERC-CoG-2014 CODOVIREVOL, Grant agreement 647916) and developed during my first postdoctoral stay working for the CNRS at the "Infectious Diseases and Vectors: Ecology, Genetics, Evolution and Control" (MIVEGEC) institute.
Experimental evolution of genome architecture in a plant RNA virus (PhD thesis)
The evolution of genome architecture in viruses can be grossly divided into three sorts of processes. First, the decrease of genome complexity, for example, the deletion of a redundant gene or regulatory sequence, which results in a reduction of genome size. Second, the increase in genome complexity, e.g. horizontal gene transfer (HGT), gene duplication, or de novo evolution of genes, which result in an increase in genome size. Third, the reshuffling of existing elements without any duplication events, which does not result in a change of genome size, but does result in a change of gene order. For my PhD thesis, I investigated these three processes of genome architecture using Tobacco etch virus (TEV), a plant RNA virus, as a model system. Molecular cloning was used to generate changes in the TEV genome. These changes consisted of changes in gene order, gene duplication and the insertion of both functional and non-functional sequences. Subsequently, experimental evolution in combination with accumulation and competitive fitness experiments, and NGS sequencing was used to asses the adaptation and fitness effects of the altered TEV genomes.

During my work in this project, I explored the multiple barriers that exist to the emergence new genome architectures in small RNA viruses. The factors that constrain or promote gene-order diversity are largely unknown, although the regulation of gene expression is one important constraint for viruses. Therefore, I investigated why gene order is conserved for a positive-strand RNA virus encoding a single polyprotein in the context of its authentic multicellular host (the plant species Nicotiana tabacum). Initially, we identified the most plausible trajectory by which alternative gene orders could evolve. Subsequently, we studied the accessibility of key steps along this evolutionary trajectory by constructing two virus intermediates: (1) duplication of a gene followed by (2) loss of the ancestral gene. We identified five barriers to the evolution of alternative gene orders. First, the number of viable positions for reordering is limited. Second, the within-host fitness of viruses with gene duplications is low compared to the wild-type virus. Third, after duplication, the ancestral gene copy is always maintained and never the duplicated one. Fourth, viruses with an alternative gene order have even lower fitness than viruses with gene duplications. Fifth, after more than half a year of evolution in isolation, viruses with an alternative gene order are still vastly inferior to the wild-type virus. Our results show that all steps along plausible evolutionary trajectories to alternative gene orders are highly unlikely (Willemsen et. al. 2016 Genetics). Hence, the inaccessibility of these trajectories probably contributes to the conservation of gene order in present-day viruses.
The factors constraining the maintenance of redundant sequences in present-day RNA virus genomes are not well known. I aimed to better understand the stability of genetic redundancy and how viruses evolve smaller genomes by removing this redundancy. The stability and fitness costs of genetic redundancy were measured, by experimentally evolving TEV variants containing potentially beneficial gene duplications (Willemsen et. al. 2016 GBE). We found that all gene duplication events resulted in a loss of viability or in a significant reduction in viral fitness. Moreover, upon analyzing the genomes of the evolved viruses, we always observed the deletion of the duplicated gene copy and maintenance of the ancestral copy. Interestingly, there were clear differences in the deletion dynamics of the duplicated gene associated with the passage duration and the size and position of the duplicated copy. Based on the experimental data, we developed a mathematical model to characterize the stability of genetically redundant sequences, and showed that fitness effects are not enough to predict genomic stability. A context-dependent recombination rate is also required, with the context being the duplicated gene and its position. Our results therefore demonstrate experimentally the deleterious nature of gene duplications in RNA viruses. Beside previously described constraints on genome size, we identified additional factors that reduce the likelihood of the maintenance of duplicated genes.
HGT is pervasive in viruses and thought to be a key mechanism in their evolution. On the other hand, strong selective constraints against increasing genome size are an impediment for HGT, rapidly purging horizontally transferred sequences and thereby potentially hindering evolutionary innovation. Here, I experimentally explored the evolutionary fate of an increase in genome size, in the context of HGT. First, a non-functional exogenous sequence (eGFP) was introduced in the TEV genome. By experimentally evolving TEV carrying eGFP (a green fluorescent protein) we have shown that carriage of this gene has a high fitness cost and its loss serves as a real-time indicator of adaptation (Zwart et. al 2013). The strength of selection for a reduced genome size and the rate of pseudogenization depend on demographic conditions, where passage length plays an important role. Second, two different exogenous sequences were introduced in the TEV genome, that simulate the acquisition of a new function and the acquisition of an existing function. We found that the insertion of a gene with a new function was rapidly purged from the viral genome, restoring fitness to wild-type levels (Willemsen et. al. 2017 GBE). Conversely, the insertion of gene (from another viral family) with an existing function was stably maintained and did not have a major impact on viral fitness. Moreover, we found that this inserted gene is functional in TEV, as it provides a replicative advantage when the original TEV gene with the same function is mutated. These observations suggest a potentially interesting role for HGT of short functional sequences in ameliorating evolutionary constraints on viruses, through the duplication of functions.
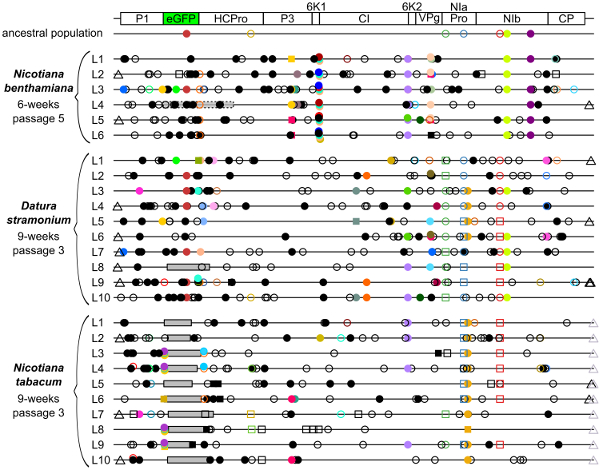
To better understand the effects of virulence and transmission on evolution, I considered whether the evolutionary patterns observed for viruses with an altered genome architecture are similar in alternative hosts. By using hosts for which TEV has a large difference in virulence, I explored how virulence affect adaptability to a new host. We used TEV carrying the eGFP fluoresent marker and evolved this virus in two semi-permissive host species, and compared the results to those obtained in the natural host. After over half a year of evolution, we sequenced the genomes of the evolved lineages and measured their fitness. During the evolution experiment, marker loss leading to viable virus variants was only observed in one lineage of the host for which the virus has low virulence (Willemsen et. al. 2017 BMC Evol Biol). This result was consistent with the observation that there was a fitness cost of eGFP in this host, while surprisingly no fitness cost was observed in the host for which the virus has high virulence. Furthermore, in both hosts we observed increases in viral fitness in few lineages, and host-specific convergent evolution at the genomic level was only found in the host for which the virus has high virulence. The results of this study exemplify that jumps between host species can be game changers for evolutionary dynamics. When considering the evolution of genome architecture, host species jumps might play a very important role, by allowing evolutionary intermediates to be competitive.
This project was financed by an EC ICT grant (FP7-ICT-2013-10 EvoEvo, Grant agreement 610427) and a John Templeton Foundation grant (JTF22371) and developed during my PhD at the Evolutionary Systems Virology Group (EvolSysVir) under the supervision of Dr. Santiago F. Elena and Mark P. Zwart.
For a general overview of the fate of genomic insertion in a plant RNA virus, see Willemsen et. al. 2018.
For a review on the stability of sequences inserted into all types of viral genomes, see Willemsen and Zwart 2019.
